
43- PATOGÉNESIS MOLECULAR DE LAS ATAXIAS ESPINOCEREBELARES.
Autores: Drs.Antoni Matilla Dueñas, Robert Goold, y Paola Giunti. (Institute of Child Health, and Department of Molecular Neuroscience, Institute of Neurology, University College, de Londres).
Copiado de: "Brain Advance Access", doi:10.1093/brain/awl081 Brain (2006).
Fecha de publicación: 13 de abril del 2006.
Traducción desde el inglés: Miguel-A. Cibrián, con revisión posterior del Dr. Antoni Matilla Dueñas.
Las ataxias espinocerebelosas autosómicas dominantes (SCA's) comprenden un grupo de enfermedades neurodegenerativas, clínica y genéticamente heterogéneas, caracterizadas por la pérdida de equilibrio y coordinación motora, debido a disfunción progresiva del cerebelo y sus conexiones aferentes y eferentes. A pesar de los bien descritos fenotipos clínicos y patológicos, aún se entienden poco los eventos moleculares y celulares neurodegenerativos que las acompañan.
La evidencia apunta hacia papeles etiológicos, de interferencia con la regulación transcripcional, agregación y eliminación de proteínas, el sistema de la ubiquitina-proteasoma, y alteraciones de homeostasis del calcio intracelular, en la pérdida neuronal observada durante el proceso neurodegenerativo. Sin embargo, también han sido identificadas nuevas rutas moleculares que podrían irrumpir durante la progresión de la enfermedad. Estas vías podrían actuar independientemente o, más probablemente, interactuar con otras, activando la acumulación del deterioro celular que en el futuro lleva al trastorno y, finalmente, a la muerte de las neuronas a través de una serie de eventos múltiples. Esto sugiere que pudieran ser objetivos necesarios tratar terapéuticamente en varias vías simultáneamente para prevenir la neurodegeneración y preservar la función neuronal. Entender cómo la disregulación de estas vías media en la progresión de la enfermedad, está llevando al establecimiento de estrategias terapéuticas eficaces, in vivo, que pueden mostrarse beneficiosas en el tratamiento de las SCA's. En este artículo, repasamos la más reciente evidencia respecto a los procesos moleculares propuestos para la patogénesis de las ataxias espinocerebelosas de herencia dominante y las estrategias terapéuticas actuales que se están llevando a cabo.
Abreviaturas: ADCA = ataxias espinocerebelosas autosómicas dominantes. CAG = secuencia de ADN que codifica el aminoácido glutamina. DRPLA = atrofia dentatorubropalidoluisiana. ER = retículo endoplásmico. FGF14 = factor de crecimiento de fibroblastos 14. HDACs = histonas deacetilasas. KCNC3 = canal de voltaje del potasio. PP2 = proteína fosfatasa 2. PRKCG = proteína kinasa C gamma. Q = aminoácido glutamina. SCA = ataxia espinocerebelosa. SPTBN2 = espectrina beta-III. TBP = proteína de union de la secuencia TATA en el ADN. UPS = sistema ubiquitina-proteasoma.
Introducción:
Las ataxias espinocerebelosas autosómicas dominantes (SCA's) son un grupo complejo de desórdenes neurodegenerativos caracterizados por ataxia cerebelosa progresiva al caminar y en los miembros, asociada variablemente con oftalmoplegia, signos piramidales y extrapiramidales, demencia, retinopatía pigmentaria, y neuropatía periférica (Zoghbi, 2000). El inicio de la enfermedad normalmente tiene lugar entre los 30 y 50 años de edad, aunque también se han detectado algunas formas con inicio temprano, en la niñez, y con inicio tardío, en décadas posteriores a los 60 años. La prognosis es variable, dependiendo de la causa subyacente del subtipo de ataxia espinocerebelosa. Los datos epidemiológicos indican que las SCA's podrían ser más frecuentes de lo estimado en un primer momento, con prevalencias de hasta 5 a 7 por 100.000 habitantes en algunas poblaciones (van de Warrenburg et al., 2002; Craig et al., 2004). La prevalencia es similar a la de otras enfermedades neurodegenerativas motoras, como la enfermedad de Huntington o enfermedades de las motoneuronas. Los efectos de una mutación fundadora parecen contribuir a la variable prevalencia detectada en subtipos específicos de SCA en distintas poblaciones.
En la era pregenómica, las SCA's han sido particularmente polémicas en lo referido a su nomenclatura y clasificación. Harding, en un primer lugar, propuso la clasificación de las ataxias cerebelosas autosómicas dominantes (ADCA's) en base a los síntomas clínicos, y diferenció este conjunto de desórdenes en tres grupos principales (Tabla 1) (Harding, 1993).

Es importante anotar que la clasificación de Harding no ha quedado obsoleta por la clasificación genética: y sigue siendo válida como pauta en la práctica clínica y para priorizar las pruebas genéticas para el diagnóstico. Las ADCA tipo I se caracterizan variablemente por ataxia al caminar asociada con oftalmoplegia, signos piramidales y extrapiramidales, deterioro cognitivo, atrofia óptica, o neuropatía periférica. Los rasgos clínicos en este grupo de ataxia están causados por una combinación de degeneración del cerebelo, ganglios basales, corteza cerebral, nervio óptico, sistemas ponto-medulares, tractos espinales, o nervios periféricos. Las ADCA tipo II se distinguen de las ADCA tipo I por la presencia de retinopatía pigmentaria. Un tercer grupo, ADCA tipo III, incluye aquellas ataxias cerebelosas relativamente puras, en las cuales, el proceso degenerativo se limita al cerebelo. Las ADCA's I y III son claramente heterogéneas genéticamente, y al menos dos genes diferentes se han asociado con las ADCA II (Tabla 1) (Giunti et al., 1999). Aunque la etiología de las SCA's aún no está bien entendida, los análisis genéticos, los datos epidemiológicos, las investigaciones neuropatológicas, y los nuevos modelos animales experimentales, están proporcionando nuevas visiones importantes de los mecanismos patogénicos. Por lo menos 28 locus distintos son responsables de las distintas formas de SCA (Tabla 2). Algunos subtipos de SCA, incluidas las SCA's 1, 2, 3, 6, 7, 17, y la atrofia dentatorubropalidoluisina (DRPLA), están causadas por una expansión CAG (secuencia de ADN que codifica el aminoácido glutamina) localizada dentro de la región codificante de genes específicos, con lo que se producen expansiones de poliglutamina anormalmente largas (poliQ) en las proteínas codificadas, llamadas ataxinas 1, 2 y 3, alfa 1A-voltaje-dependiente del canal del calcio, ataxina 7, proteína de union a la secuencia TATA (TBP), y atrofina 1, respectivamente. Estas SCA's muestran, como rasgos comunes, la neurodegeneración progresiva de subconjuntos neuronales en distintas áreas del cerebro y la formación de poliQ conteniendo proteínas agregadas formando las típicas inclusiones nucleares o citoplásmicas (Zoghbi y Orr, 2000). La edad de inicio y la severidad de los síntomas de la enfermedad se correlacionan inversamente con la longitud de las repeticiones de glutamina. Un segundo grupo de SCA's, incluidas las SCA's 8, 10 y 12, son causadas por una repetición de la expansión localizada fuera de la región codificante de los genes de la enfermedad, llevando a la desregulación de la expresión del gen (Tabla 2) (Holmes et al., 1999; Koob et al., 1999; Matsuura et al., 2000).

Mientras los mecanismos moleculares subyacentes en las SCA's 8 y 10 son inciertos, la SCA12 parece ser causada por disregulación de la actividad de la proteína de la enzima crucial fosfatasa 2 (PP2, anteriormente llamada PP2A) en las células cerebelosas de Purkinje. Mecanismos diferentes causan la ataxia cerebelosa y la neurodegeneración en las SCA's 5, 13, 14 y 27, en las cuales las alteraciones en la composición del aminoácido en la proteína espectrina beta-III (SPTBN2) (Ikeda et al., 2006), canal de potasio KCNC3 (Waters et al., 2006), proteína kinasa C (PRKCG) (Chen et al., 2003a; Yabe et al., 2003) y factor de crecimiento de fibroblastos 14 (FGF14) (van Swieten et al., 2003), respectivamente, producen los síntomas de la enfermedad en estos cuatro subtipos de SCA. En el resto de las SCA's, los genes y, por consiguiente, las mutaciones permanecen sin identificar y caracterizar. Entender los mecanismos patogenéticos de la neurodegeneración en las ataxias espinocerebelosas deben llevar a la identificación de potenciales dianas terapéuticas y, finalmente, deben facilitar el descubrimiento de medicamentos efectivos.
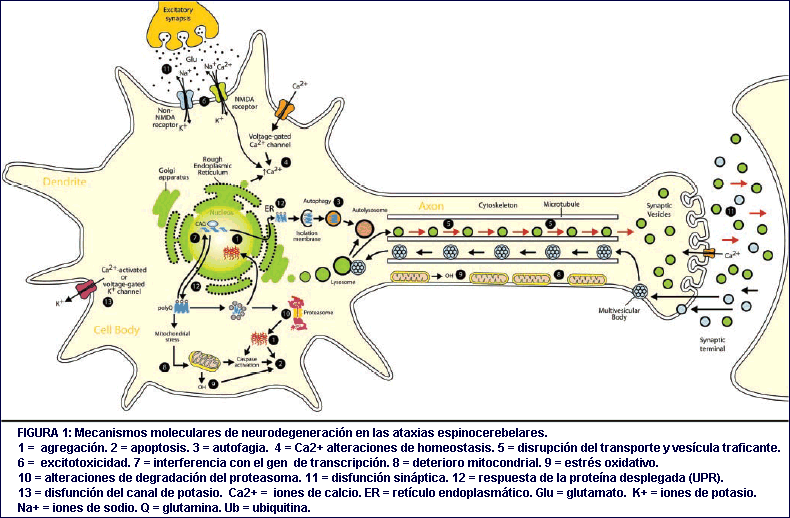
Esta revisión resalta la evidencia científica para las mayores vías que llevan a la neurodegeneración en las SCA's autosómicas dominantes, desde el análisis genético, en los estudios in vitro, en los modelos animales experimentales, y los estudios cerebrales post-mortem (Figura 1). Debatimos la convergencia de estas vías en la identificación de potenciales dianas moleculares que podrían usarse para desarrollar tratamientos terapéuticos eficaces (Tabla 3).

Neurotoxicidad de la poliglutamina: agregación de la proteína, mal plegada, estabilidad y eliminación:
Siete subtipos de ataxias espinocerebelosas incluidas las SCA's 1, 2, 3/Enfermedad de Machado-Joseph, 6, 7, 17, y DRPLA, están causadas por la expansión de secuencias repetidas CAG en los genes específicos, produciendo fragmentos anormalmente largos de poliQ en las proteínas codificadas (Zoghbi y Orr, 2000). Probablemente, está involucrada en estos SCA's una vía patogénica similar, puesto que cada una de estas expansiones repetidas CAG codifica poliglutamina y el umbral patogénico para la enfermedad es el mismo, alrededor de 40 copias de la repetición en la mayoría de los diferentes subtipos. Se supone que estas SCA's tienen en común los efectos tóxicos producidos por la proteína con poliglutamina que incluyen agregación y deposición de proteínas mal plegadas que llevan a la disfunción neuronal y finalmente a la muerte celular.
Las proteínas con expansiones expandidas de poliglutamina parecen asumir una configuración anormal que resulta en la formación y deposición de agregados de poliglutamina formando las características inclusiones nucleares o citoplásmicas que son peculiaridades neuropatológicas en estas enfermedades (Ross y Poirier, 2004). Estas inclusiones contienen componentes celulares como la ubiquitina, el proteasoma, HSP70 y factores de transcripción (Cummings et al., 1998; McCampbell et al., 2000; Schmidt et al., 2002). O bien, la toxicidad es un resultado directo del agregado, o resulta a partir de las estructuras intermediarias formadas durante el proceso de agregación. No obstante, bloquear la agregación ha sido un acercamiento intentado para minimizar la toxicidad. Las proteínas expandidas de poliglutamina forman agregados proteinaceos fibrilares mucho más rápidamente que las proteínas normales (Chow et al., 2004). Por consiguiente, un posible mecanismo para la formación de agregados por la proteína mutada sería debido a la alteración de estabilidad del estatus nativo por la poliglutamina expandida, y así, llevando a la formación y acumulación de agregados que producen fibrilización. Este fenómeno podría considerarse para la edad más cercana al inicio y la severidad más alta de los síntomas de la enfermedad observada cuando las ataxinas mutadas contienen números más amplios de glutaminas. Enigmáticamente, a pesar del hecho de que la mayoría de las proteínas asociadas con enfermedades neurodegenerativas espinocerebelosas se expresan sistemáticamente, la citotoxicidad resultante parece estar restringida a algunos subtipos neuronales del Sistema Nervioso Central (SNC). Las condiciones celulares selectivas y las interacciones de la proteína con proteínas específicas podrían conferir condiciones insolubles locales llevando a la oligomerización y fibrilización en las neuronas vulnerables.
La relación entre la neurodegeneración en SCA y el sistema proteasoma ubiquitina-dependiente (UPS):
La maquinaria celular principal para degradar las proteínas aberrantemente plegadas, se evidencia por los resultados consistentes de agregados de proteína ubiquitina en los estudios neuropatológicos (Ross y Poirier, 2004). Además, hay evidencia de que algunas ataxinas, como las ataxinas 1, 3, y 7, son susceptibles de ser ubiquitinadas y son objetivo del proteasoma para la degradación y eliminación (Cummings et al., 1999; Matilla et al., 2001; Chai et al., 2004). La proteína mal plegada ejercida por la poliglutamina expandida llevaría a dificultades en el reconocimiento y proceso de degradación por parte del proteasoma, y la eliminación subsecuente de las proteínas mutadas. La amplia evidencia indica que las expansiones de la poliglutamina y los agregados también pueden disregular la función del UPS. Por ejemplo, la poliglutamina-expandida en ataxina 1 parece disminuir la actividad del proteasoma en células en cultivo (Park et al., 2005). En SCA3, la ataxina 3 es una ubiquitina-específica, cisteina proteasa, que se asocia con el proteasoma (Chai et al., 2004; Nicastro et al., 2005), y deubiquitina proteínas por el ligado de las cadenas del poliubiquitina a través de varios motivos de interacción con ubiquitina (UIMs) dentro del dominio Josephin, el cual se localiza cerca del dominio polyQ. La elucidación de la estructura molecular del dominio Josephin dentro de la ataxina 3 ha llevado a la propuesta de un modelo para el reconocimiento de interacciones y formación de complejos estables con HHR23B localizado en la misma superficie involucrada en la interacción con los dominios UBA, el S5a y los sitios de unión a poliUb y al proteasoma (Nicastro et al., 2005). Estas observaciones proporcionan un enlace entre la ataxina 3 y el UPS que es de particular relevancia para las enfermedades neurodegenerativas. Las poliglutaminas expandidas dentro de la ataxina 3 podrían alterar su función normal y producir disrupciones funcionales de la vía UPS. Alternativamente, la reducción de la actividad del proteasoma puede ser el resultado de la degradación por el sistema caspasa-dependiente de subunidades del proteasoma que inducen fenómenos de apoptosis (Sun et al., 2004). Puesto que el UPS juega un papel prominente en la detoxificación y marcado de las proteínas dañadas para la degradación, el fallo de este sistema podría llevar a una acumulación anormal de una variedad de proteínas tóxicas, incluyendo las que contienen poliglutamina, que, de lo contrario, hubieran sido degradadas, llevando finalmente a la disfunción y/o muerte neuronal.
Las chaperonas moleculares, proteínas que pueden reparar o facilitar la degradación del proteasoma de proteínas anormalmente plegadas, pueden jugar un papel en las SCA's ya que los agregados que se encuentran en el tejido humano post-mortem contienen chaperonas (Ross y Poirier, 2004). Esta evidencia indica que los mecanismos de supervivencia celular son mediados por el retículo endoplásmico (ER), chaperonas, y la respuesta a la proteína desplegada (UPR) activada durante la neurodegeneración en las ataxias espinocerebelosas. La presencia de proteínas desplegadas en el ER puede causar estrés ER o un desequilibrio entre la carga de proteínas mal plegadas y la capacidad del ER para procesarlas. Para restaurar la homeostasis del ER, las neuronas activan la respuesta estrés del ER o la respuesta UPR, desencandenando la activación transcripcional de genes codificando chaperonas. Consistente con esta hipótesis, la sobrexpresión experimental de chaperonas moleculares modula la formación de proteínas agregadas en las células en cultivo, ratones transgénicos, y moscas de la fruta, disminuyendo la toxicidad de las expansiones de glutamina (Cummings et al., 1998; Cummings et al., 2001; Bonini, 2002).
żCómo modulan la toxicidad de la proteína las chaperonas?:
Un posible mecanismo podría ser estabilizando la estructura monomérica proteica mal plegada. Las chaperonas podrían prevenir la transición intramolecular, que da lugar a oligomeros esféricos y anulares y, simultáneamente, estabilizar una estructura que promueve la formación de los cuerpos de inclusión (Sakahira et al., 2002). Alternativamente, mediante la interacción con la proteína de la enfermedad, las chaperonas podrían prevenir las interacciones anormales con otras proteínas que causan toxicidad celular.
Las proteínas que continúan mal plegadas son degradadas principalmente por el sistema ubiquitina-proteasoma, pero también por el sistema fagosoma-lisosoma, un proceso celular conocido como autofagia, el cual contribuye a la producción rutinaria de componentes citoplásmicos (Shintani y Klionsky, 2004). Paradójicamente, la autofagia es usada para proteger a las células, pero también contribuye a la muerte celular. La acumulación de vesículas (cuerpos) autofágicos ha sido observada en muchas enfermedades neurodegenerativas con asociados y agregados mal plegados. La reciente evidencia ha mostrado que la autofagia juega un papel esencial en la liquidación celular de proteínas agregadas tóxicas en células cultivadas de modelos de poliglutamina-expandida ataxina 1 que media en la neurodegeneración (Iwata et al., 2005). El mecanismo preciso por el cual la ataxina 1 agregada mutada es capturada por los autofagosomas es desconocido, pero parece ser que la inhibición de mTOR, un regulador negativo de eliminación autofágica, induce a la autofagia y reduce la toxicidad de las expansiones de poliglutamina en los modelos de mosca y murino de neurodegeneración (Ravikumar et al., 2004). Este estudio ha establecido eficazmente que la autofagia degrada los agregados citoplásmicos insolubles, y apunta a una posible estrategia terapéutica modulando el proceso autofágico para promover la eliminación de las proteínas agregadas en la enfermedad. Es importante anotar que la autofagia también pueden inducir a la muerte celular en neuronas que acumulan proteínas agregadas, en cierto modo eso produce una condición patológica, aunque en la actualidad los mecanismos moleculares son desconocidos.
En 1993, fue propuesta una hipótesis en la cual las inclusiones neuronales de proteínas inducidas por las poliglutaminas expandidas son estabilizadas por isopéptidos covalentemente unidos introducidos por la transglutaminasa (Green, 1993; repasado en Hoffner y Djian, 2005). Fue postulado que, como resultado de la reiteración excesiva de glutamina, las proteínas mutadas se convertirían en substratos de la transglutaminasa. De hecho, en experimentos in vitro se demostró posteriormente que una glutamina repetida, si es expuesta en la superficie de una proteína, debe formar agregados covalentemente unidos en presencia de cualquier actividad de transglutaminasa activada por Ca2+. Mientras que no ha sido fácil examinar la actividad de la transglutaminasa, el tratamiento de células en cultivo llevando atrofina 1 truncada en presencia del inhibidor trangluminasa cistamina reduce la formación de agregados y la muerte celular (Igarashi et al., 1998). Estas observaciones proporcionan argumentos en favor de la hipótesis de la transglutaminasa.
Alteraciones de la homeostasis del calcio:
La alteración de la homeostasis del calcio juega un papel central en apoptosis, y la sobrecarga o perturbación de la compartimentalización intracellular del catión Ca2+ son potencialmente citotóxicas (Orrenius et al., 2003). El almacenamiento y la señalización del calcio, así como el plegando, modificado, y procesado de proteínas recientemente sintetizadas están entre las funciones principales del ER en las células de mamíferos. Las perturbaciones en cualquiera de estas funciones pueden llevar al llamado estrés del ER. Ambos, la sobrecarga de Ca2+ y los niveles de Ca2+ del ER o las alteraciones en el sistema de trasporte Ca2+ pueden producir cambios en el plegado proteico, en la respuesta a estrés del ER, y en la activación de las vías pro-apoptóticas. La mitocondria es conocida por participar activamente en la compartimentalización intracelular del Ca2+ (Petersen, 2002). Se ha clarificado que el flujo de calcio mitocondrial integra partes del señalizador celular Ca2+. El calcio regula la permeabilización de la membrana mitocondrial exterior (OMM) y, por consiguiente, la descarga de proteínas pro-apoptóticas mitocondriales al citoplasma, como el citocromo C y el factor inductor de apoptosis (AIF). Éste se acopla a un proceso de respuesta al estrés conocido como la transición de la permeabilidad de la membrana interna.
Fuerte evidencia apunta a anomalías de la señalización del calcio neuronal en la neurodegeneración de las ataxias espinocerebelosas. Las células cerebelosas de Purkinje parecen ser particularmente sensibles a los flujos en los niveles del calcio intracelular, que puede resultar desde diferentes niveles como por ejemplo por reducción de actividad de las chaperonas y el estrés del ER. Los niveles de expresión de varios genes neuronales, abundantemente expresados en las células de Purkinje, involucrados en la señalización del calcio o en la homeostasis están disminuidos en el cerebelo de los ratones mutados modelo de SCA1 antes de tener lugar el detectable deterioro motor o los signos patológicos (Lin et al., 2000; Serra et al., 2004). Esto hace pensar en un mayor papel de disrupción de la homeostasis del calcio en las células cerebelosas de Purkinje, que podrían estar altamente involucradas en el proceso patogénico y/o supervivencia de estas células en la SCA1.
En SCA6, la degeneración de la células de Purkinje es asociada con las expansiones de poliglutamina dentro del gen CACNA1A (Zhuchenko et al., 1997). El gen CACNA1A codifica un subunidad alfa (2.1), anteriormente llamada [alpha]1A, la cual es la mayor subunidad del CaV2.1, canal del calcio tipo-P/Q voltaje-dependiente (Pietrobon, 2002). Los canales del calcio voltaje-dependientes están compuestos por subunidades adicionales beta's y gamma's. Las subunidades alfa son glicoproteínas de membrana de 2400 aminoácidos en longitud, en los cuales la estructura primaria predice la presencia de cuatro dominios homólogos, cada uno consiste en seis dominios de transmembrana y un dominio P-loop de formación del poro. Los canales del calcio tipo/PQ de activación de alto-voltaje se hallan principalmente en las neuronas y expresados a altos niveles en las células gránulo y células de Purkinje de la corteza cerebelosa. Se cree que su papel principal está en la transmisión sináptica. Queda por ser demostrado si la acción del producto del gen mutado es perturbar la función del cauce del calcio, servir como diana para las transaminasas, o bien ligar proteínas de unión al ADN. Interesantemente, tres enfermedades alélicas están causadas por tipos diferentes de mutaciones en el gen de CACNA1A, incluyendo la SCA6, la ataxia episódica tipo 2, y la migraña hemipléjica familiar (Mantuano et al., 2003). Una forma de ataxia cerebelosa asociada con una mutación de G293R en el P-loop del primer dominio del canal CaV2.1 tiene un fenotipo muy similar al de la SCA6 asociada con expansiones de repetición CAG. Como las mutaciones puntuales en el gen CACNA1A no actúan vía hipotéticos mecanismos de unión al ADN ni tienen efectos en la transaminación, una alteración de la función del cauce de calcio podría ser un posible efecto causativo de los alelos patogénicos. Las expansiones de poliglutamina en SCA6 podrían interferir con el canal Ca2+ para reducir la entrada Ca2+ y dañar la función del mutado canal Ca2+, volviéndole incapaz de prevenir la muerte celular (Matsuyama et al., 2004). Aún se necesitan más estudios electrofisiológicos para clarificar los mecanismos moleculares subyacentes de la patogénesis de la enfermedad en la SCA6 y en otros tipos de ataxia relacionados.
Estrés mitocondrial y apoptosis:
Múltiples líneas de evidencia sugieren que la muerte neuronal en las ataxias espinocerebelosas son el resultado de la activación directa de vías apoptóticas (Lipinski y Yuan, 2004). Además del papel de la mitocondria en la homeostasis del calcio intracelular, como se ha mencionado anteriormente, la muerte celular causada por expansiones de poliglutamina en neuronas cerebelosas por las ataxinas 3 y 7, en SCA3 y SCA7, respectivamente, es precedida por el reclutamiento de las caspasas en los agregados polyQ. Esto es seguido por la activación de las caspasas 3 y 9, y de las vías apoptóticas mitocondriales mediadas por miembros de la familia de Bcl-2, como Bax y Bcl-x(L) (Chou et al., 2006; Wang et al., 2006). Ambos factores son conocidos componentes de la apoptosis neuronal, regulando la liberación mitocondrial de citocromo C y Smac/DIABLO. Consistente con estas observaciones, la ataxina 7 mutada, en SCA7 induce la translocación de citocromo C y Smac/DIABLO al citosol precedido por la activación de las caspasas 9 y 3. Esto sugiere que la ataxina 7 mutada causa muerte apoptótica de las neuronas cerebelosas promoviendo la liberación mitocondrial de citocromo C y de Smac/DIABLO. Alternativamente, las vías pro-apoptóticas podrían ser activadas o por el desplazamiento de factores perjudiciales secuestrados por poliglutaminas expandidas o a través de los mecanismos anormales de activación de caspasas. En todo caso, las proteínas tóxicas pueden promover la disfunción mitocondrial e incrementar la producción de radicales libres asociados con el daño oxidativo, y concentración y la utilización anormal de la energía metabólica. La amplia evidencia de la disfunción mitocondrial resultando o contribuyendo a la ataxia se ejemplifica por enfermedades causadas por mutaciones en polimerasa gamma, resultando en predominante disfunción/atrofia cerebelosa. Aunque relevantes, estos mecanismos de toxicidad no explican la vulnerabilidad selectiva de distintos tipos de células y regiones del cerebro en las neurodegeneraciones espinocerebelosas.
Vías emergentes:
La mayoría de las vías que conducen a la toxicidad por proteínas enfermas en las ataxias espinocerebelosas son todavía desconocidas. La evidencia creciente revela que la expresión nuclear de proteínas de poliglutamina expandida es un paso esencial en la patogénesis molecular, y que la disregulación transcripcional resultante causada por ataxinas conteniendo poliglutaminas expandidas produce disfunción y muerte neuronal. La interferencia con la expresión del gen tendría lugar por los efectos de la mutación ejercidos en la interacción transcripcional con las proteínas reguladoras. Por ejemplo, la ataxina 1 interactúa con el represor transcripcional SMRT (silenciador mediador de los receptores de las hormonas retinoides y tiroides) a través del dominio AXH en Atxn1 (Tsai et al., 2004). Puesto que SMRT regula la expresión de los receptores de las hormonas retinoidea y tiroidea, es factible que la forma mutada de ataxina 1 podría ejercer efectos deletéreos en las vías transcripcionales reguladas por SMRT. Similarmente, las ataxinas 3 y 7, y los productos génicos de SCA17 y DRPLA, TBP y atrofina 1, respectivamente, están directamente involucrados en la transcripción como componentes de complejos reguladores transcripcionales (Shimohata et al., 2000; Li et al., 2002; Zhang et al., 2002; Helmlinger et al., 2004).
Sin embargo, los genes y vías reguladas por estos factores permanecen desconocidos. La disregulación transcripcional podría considerarse para la degeneración de células específicas observada en las ataxias espinocerebelosas, a través de factores, por expansiones de la glutamina dentro de las proteínas mutadas cuya expresión es confinada a las células que degeneran en estas condiciones. Por consiguiente, se volverían indisponibles para realizar sus deberes celulares normales, con consecuencias potencialmente letales.
En SCA1, el secuenstramiento subcelular de la proteína nuclear acídica 32 (Anp32a/Lanp) (Matilla et al., 1997), PQBP-1 (Okazawa et al., 2002), Boat (Mizutani et al., 2005) y Gfi-1/Senseless (Tsuda et al., 2005) podría conducir a la neurodegeneración cerebelosa selectiva dañando la transcripción y disregulando expresión génica. Importantemente, Anp32a/Lanp es un potente regulador específico de la actividad enzimática de PP2 y su expresión se confina predominantemente a las células cerebelosas de Purkinje (Matilla y Radrizzani, 2005). Por consiguiente, la disregulación de la actividad de PP2 es un probable mecanismo que subyace en la neurodegeneración cerebelosa de la SCA1. De acuerdo con esto, en SCA1 y SCA12, la muerte celular podría ser provocada por mecanismos patogénicos similares. De forma similar, la inactivación de la transcripción en tejido-específico por el factor CRX en SCA7 debido a la expansión de glutamina en la ataxina 7 mutada podría contribuir a la distrofia de los conos retinales observada en ratones transgénicos y pacientes de SCA7 (Spada et al., 2001). En particular, un grupo de proteínas nucleares a las que ligan y cuya actividad puede ser alterada por las ataxinas mutadas son las histonas acetiltransferasas (HATs).
Las HATs son responsables de la acetilación de histonas y otras proteínas, y este fenómeno regula la función de la proteína modificada (Kouzarides, 2000). Las HATs también regulan procesos celulares a diferentes niveles, modifican la competencia transcripcional de los cromosomas, se involucran en la regulación temporal del interruptor de actividad de la proteína, y en la activación/inactivación proteica. El equilibrio alterado entre la acetilación/deacetilazión proteica puede ser un proceso importante que contribuyen a la patogénesis inducida por proteínas mutadas que contienen poliglutaminas expandidas (Steffan et al., 2001). Importantemente, la regulación de este equilibrio es posible por la reducción genética o farmacológica del grupo de enzimas contrario, es decir, las histonas deacetilasas (HDACs). Esto está llevando a nuevas estrategias terapéuticas con inhibidores de la histona desacetilasa (HDACis) para tratar la neurodegeneración (McCampbell et al., 2001; Steffan et al., 2001; Hockly et al., 2003).
Las líneas de evidencia surgidas son de alta importancia para entender la función normal de las proteínas de la enfermedad asociadas con las ataxias espinocerebelosas y desentramar los mecanismos moleculares que hay bajo los efectos perjudiciales producidos por las mutaciones. En SCA1, la expansión del tracto de poliglutamina o la expresión nuclear por sí sola no son suficientes para causar la enfermedad. Parece ser que la fosforilación de un específico residuo de serina en la ataxina 1 por la proteína kinasa B/Akt juega un papel crucial modulando la habilidad de la forma mutada de la ataxina 1 para inducir la neurodegeneración influyendo en sus interacciones biológicas con otras proteínas y formando inclusiones nucleares (Chen et al., 2003b). Por consiguiente, es probable que el contexto de la proteína y de las proteínas celulares con las cuales normalmente interactúa la ataxina 1 sean importantes en el proceso de la enfermedad, indicando que, por ejemplo, bloquear los eventos de fosforilación podría ser un tratamiento terapéutico viable. En apoyo de esto, el contexto de la proteína parece ser un factor modificador de la edad de inicio de las degeneraciones espinocerebelosas (van de Warrenburg et al., 2005). En SCA2, la ataxina 2 contiene un dominio Lsm ARN-obligatorio caracterizado por un motivo de secuencia conservado que consiste en dos segmentos cortos conocidos como Sm1 y Sm2, los cuales están separados por un espaciador variable (Albrecht et al., 2004). Los dominios de proteínas Lsm están involucrados en una variedad de eventos del proceso ARN, incluida la modificación de ARN, uniendo, cortando, y degradando el pre-mRNA, y algunos de ellos también son componentes importantes del espliceosoma, pequeños complejos nucleares de ribonucleoproteína (snRNPs) (He y Parker, 2000). Interesantemente, la ataxina 2 interactúa con A2BP1 (ataxina 2 proteína ligada 1) (Shibata et al., 2000) cuyo ARN-ligado en el homólogo del gusano Caenorhabditis elegans, fox-1, regula tejidos específicos alternativos (Jin et al., 2003). Descifrando los mecanismos por los que la ataxina 2 regula ligamientos alternativos, debe proporcionar intuiciones dentro de las vías desreguladoras durante la progresión de la enfermedad llevando a la identificación de potenciales dianas terapéuticas.
La neurotoxicidad mediante el ARN ha sido implicada en la patogénesis de la SCA8. Parece que el gen de SCA8 se transcribe como una parte de un ARN no-traducido que se solapa con la transcripción y sitios de inicio de traducción y la primera unión del empalme de KLHL1, un gen que codifica una proteína con similitudes estructurales a una familia de factores involucrados en la organización del esqueleto celular (Koob et al., 1999). Puesto que el gen SCA8 funciona como un gen regulador, ha sido propuesto que un mecanismo de ganancia de función de ARN podría causar la neurodegeneración en SCA8, como es el caso para la distrofia miotónica (DM) (Philips et al., 1998; Liquori et al., 2001). Análisis genéticos en moscas ha revelado que las mutaciones en los genes "staufen, músculo-blind, split ends y CG3249" modulan la neurodegeneración (Mutsuddi et al., 2004). Estas observaciones sugieren que las expansiones CUG en SCA8 pudieran alterar las asociaciones con específicas proteínas de union al ARN. Es importante anotar que el defecto molecular en la SCA8 está sujeto a controversias, puesto que en las expansiones en el gen SCA8 no se detectan exclusivamente en los pacientes con ataxia espinocerebelosa (Worth et al., 2000; Corral et al., 2005).
La SCA10 es causada, singularmente, por una repetición intrónica pentanucleotídica ATTCT dentro del gen E46L, ahora designado ATXN10 (Matsuura et al., 2000). La proteína E46L, expresada ampliamente a lo largo del cerebro, contiene dos dominios armadillos (brazos) repetidos, e interactúa con la proteína heterotrimérica de unión a GTP (proteína-G) (Lin y Ashizawa, 2005). Ha sido propuesto que E46L refuerza la señalización por la proteína-G heterotrimérica para mediar la formación de neuritis. E46L pertenece a la familia de proteínas en las cuales el brazo repite las interacciones proteína-proteína para modular un vasto número de procesos celulares, incluyendo señalizaciones intracelulares, regulación citoesquelética, transporte nuclear, y regulación de la expresión del gen durante el desarrollo y a lo largo de la vida adulta (Coates, 2003). Por consiguiente, E46L podría regular procesos celulares importantes a través de la mediación de la proteína-G de señalización intracelular. Una repetición expandida de trinucleótido CAG localizada en la región promotora del gen codifica una subunidad de PP2 (anteriormente 2A) (PPP2R2B) ha sido asociado con SCA12 (Holmes et al., 1999).
La neurodegeneración en SCA12 está confinada al cerebelo. La fosfatasa serina/treonina PP2 regula una amplia serie de procesos celulares, incluidos el crecimiento y diferenciación celular, replicación del ADN, morfogénesis celular, depresión sináptica a largo término, y apoptosis (Sontag, 2001). Se ha propuesto que las expansiones de la repetición en SCA12 pueden alterar los niveles de expresión de una variante denominada B beta 1 de la subunidad regulatoria de PP2, PPP2R2B, con influencia en la expresión debido a la eficiencia del promotor, con consecuencias potencialmente letales. Es posible que la expansión de SCA12 también pudiera tener un efecto sobre la generación de PPP2R2B, o altere la expresión del gen de algún otro modo. Hasta ahora no hay evidencia para sugerir que la repetición CAG se traduce en poliglutamina. La enzima PP2 desfosforila una diversidad de kinasas, incluida la PKB/Akt y PKCs, que median en los procesos neurodegenerativos en las SCA's 1 y 14, respectivamente (Chen et al., 2003a, b). Interesantemente, la disregulación de PP2 promueve la hiperfosforilación de tau, desestabliza el microtúbulo, modifica la estructura de la sinapsis, y contribuye a la neurodegeneración en la enfermedad de Alzheimer (Lim y Lu, 2005). Y significativamente, en la enfermedad de Alzheimer los niveles de actividad del PP2 se encuentran disminuídos. Por consiguiente, la disrupción de las funciones cerebrales/cerebelosas de PP2 podrían ser una vía común en los mecanismos patogénicos en las condiciones neurodegenerativas.
Los síntomas de la enfermedad en las SCA's 14 y 27 son consecuencia de alteraciones en la composición de aminoácidos dentro de la subunidad gamma de la proteína kinasa C (PRKCG) (Chen et al., 2003, a; Yabe et al., 2003) y FGF14 (van Swieten et al., 2003), respectivamente. Estas alteraciones parecen disregular la función de la proteína y las vías celulares específicas. La proteína kinasa C, calcio-fosfolípido-dependiente, abarca una familia de enzimas que transduce la plétora de señales promoviendo la hidrólisis lipídica, regulando así una variedad de procesos celulares, como la señal membrana-receptora de transdución, control de expresión del gen y plasticidad sináptica (Newton, 2001). La PRKCG se encuentra muy expresada en el cerebro y en el cordón espinal, con expresión particularmente alta en las células de Purkinje de la corteza cerebelosa durante el desarrollo dendrítico, dónde parece actuar como un regulador negativo del crecimiento y enramado dendrítico (Schrenk et al., 2002). Los productos del gen mutado PRKCG son menos estables que los del gen normal, induciendo modelos anormales de activación, alterando la membrana y reforzando su actividad. Se especula que el fenotipo de la SCA14 es el resultado de mecanismos de ganancia-de-función en lugar de haplo-insuficiencia, porque no se ha hallado ninguna mutación que termine la pauta de lectura del gen, y los animales heterozigotos PKC-nulos son neurológicamente normales (Abeliovich et al., 1993). Sin embargo, un mecanismo dominante negativo no puede descartarse actualmente.
Las alteraciones en la estabilidad de la proteína de FGF14 son el fundamento de la neurodegeneración del cerebelo y de los ganglios basales en la SCA27 (van Swieten et al., 2003). El FGF14 es miembro de una subclase de factores de crecimiento de fibroblastos que se expresa en el desarrollo y estado adulto del sistema nervioso central, y ha sido implicado en la señalización neuronal, tráfico axonal, y función sinaptosomal (Wang et al., 2002). Estudios neurofarmacológicos en ratones han mostrado que los ratones FGF14-deficientes tienen reducidas respuestas a compuestos agonistas del neurotransmisor dopamina, lo que indica que el FGF14 es requerido para la señalización dopaminérgica por el sistema dopaminérgico estriatopallidal. Por consiguiente, la disfunción de esta vía podría considerarse para explicar la hiperexcitabilidad cortical y síntomas asociados al parkinsonismo en la SCA27.
Más recientemente, han sido identificados los defectos génicos en tres ataxias espinocerebelosas. Las delecciones en el gen de la espectrina beta-III SPTBN2 cosegregan con SCA5 en familias americanas y francesas (Ikeda et al., 2006). Parece que estas mutaciones de espectrina alteran los niveles, distribución y estabilidad de la espectrina beta-III y la asociación con el transportador de glutamato EAAT4 de las células específicas de Purkinje. En una familia alemana, las mutaciones que alteran la pauta de lectura del gen de SCA5 que co-segregan con la enfermedad en individuos afectados puede romper la habilidad de la espectrina para estabilizar el citoesqueleto. Estas observaciones sugieren que la disrupción de la señalización del glutamato y de las vesículas de tráfico celular pueden jugar un papel en los mecanismos patogénicos en la SCA5, que son vías previamente implicadas en la neurodegeneración en la SCA1, Alzheimer, y las enfermedades de Huntington y esclerosis lateral amiotrófica.
En la SCA13, las mutaciones en el exon 2 del gen del canal de potasio KCNC3 se asocian con defectos en el neurodesarrollo y fenotipos neurodegenerativos (Water et al., 2006). El KCNC3 (también conocido como Kv3.3) es un subtipo de canal de potasio expresado abundantemente en el cerebelo, y las mutaciones parecen dañar la actividad del canal, siendo consistente con un efecto dominante-negativo del alelo mutado. Parece ser que las mutaciones en el canal cambian la curva de activación en dirección negativa, cerrando el canal con retraso, y, por consiguiente, podría cambiar las características del rendimiento de las neuronas cerebelosas, a las que el canal KCNC confiere capacidad por encendido de alta frecuencia.
La substitución de un solo nucleótido en la región proximal no-traducida del gen que codifica la proteína puratrofina 1, conteniendo la proteína 4 (PLEKHG4) familia G (con dominio RhoGef), una proteína implicada en la señalización intracelular y la dinámica de actina en el aparato de Golgi, está asociada con un subtipo de ataxia espinocerebelosa caracterizada por atrofia cerebelosa pura y neurosensorial de deterioro auditivo (Ishikawa et al., 2005). Interesantemente, el gen de la puratrofina 1 se localiza en la misma región cromosomática donde se ubica el gen SCA4 (Flanigan et al., 1996). El gen de la puratrofina 1, en este momento, no puede ser excluido de estar asociado con los síntomas de la enfermedad en la SCA4.
Aunque la ataxia es el síntoma prominente, pocas mutaciones causan síndrome cerebeloso casi puro y degeneración aislada de la corteza cerebelosa. Por el contrario, la mayoría de las SCA's son desórdenes multisistémicos que no sólo muestran neurodegeneración en los tractos espinocerebelosos, sino también en los ganglios basales (Schols et al., 2000). Los síntomas de Parkinsonismo en SCA1 (Gilman et al., 1996), SCA2 (Infante et al., 2004); (Simon-Sanchez et al., 2005), SCA3/MJD (Gwinn-Hardy el al., 2001), SCA6 (Kan et al., 2005), SCA17 (Gunther et al., 2004) y SCA27 (van Swieten et al., 2003) son indicativos de un posible deterioro estriatonigral y/o de las proyecciones striatopallidales en estos subtipos de ataxia espinocerebelosa.
Se ha propuesto a las alteraciones en la neurotransmisión del glutamato sináptico como un posible mecanismo que media en la neurodegeneración. En los ratones trangénicos de SCA1, la disfunción motora precede a la muerte neuronal, demostrando que la ataxina 1 mutada no produce disrupción de función motora en SCA1, no matando células, sino afectando a algunas de las funciones celulares de Purkinje, incluidas las alteraciones en la plasticidad sináptica (Clark et al., 1997). Se sugiere como un posible mecanismo, elevar los niveles de calcio intracelular por receptores de glutamato calcio-permeable (Serra et al., 2004). La importancia de mantener la transmisión de glutamato apropiada en la sinapsis célula de Purkinje/fibra paralela para la función motora es apoyada por estudios en ratones modelos de ataxia. Por ejemplo, los ratones carentes de mGluR1 despliegan deficiencias motoras significativas, y la introducción de la expresión mGlur1 en las células de Purkinje de ratones mGluR1_/_ restaura la función motora normal (Ichise et al., 2000).
Estrategias terapéuticas:
Actualmente, en ninguna de las SCA's hay tratamiento eficaz conocido para modificar la progresión de la enfermedad o frenar la neurodegeneración relacionada con el desorden, aunque sí se han obtenido beneficios en los síntomas atáxicos en algunos ensayos terapéuticos. Un ensayo preliminar abierto con acetazolamida ha reducido temporalmente la severidad de los síntomas en la SCA6 (Yabe et al., 2001). Se han usado reguladores dopaminérgicos y anticolinérgicos para aliviar el temblor, la bradikinesia o la distonia en SCA2 y SCA3 (Buhmann et al., 2003; Nandagopal y Moorthy, 2004). La espasticidad en las SCA's podría tratarse eficazmente con baclofeno, tizanadina, o mimentina. En casos seleccionados donde otros tratamientos han fallado, la toxina botulímica se ha usado para tratar con éxito la distonia y espasticidad. El temblor de intención se ha mejorado con benzodiazepinas, bloqueadores beta, o mediante estimulación talámica crónica (Pirker et al., 2003). Los calambres musculares, a menudo presentes al inicio de la condición en las SCA's 2, 3, 7, y DRPLA se alivian con magnesio, chinina, o mexiletina. Los datos preliminares de un reciente ensayo abierto con gabapentina han mostrado mejora de los síntomas cerebelosas en pacientes con ataxia mediante el aumento de la concentración del neurotransmisor GABA en el sistema nervioso central y, así, estimulando la neurotransmisión GABAérgica (Gazulla et al., 2004).
Ensayos innovadores, como con interferencia del ARN (RNAi) con objetivo de inhibir la neurodegeneración inducida por poliglutamina, prevenir la síntesis de proteínas mal plegadas y la agregación por sobrexpresión de chaperonas moleculares mediante la regulación de la expresión de sus genes por aplicación de HDACis, está mostrando eficacia en ensayos preclínicos. En la SCA1, la inserción intracerebelosa de vectores expresando pequeños ARN's mejora la coordinación motora, restaura la morfología cerebelosa, y reduce, en ratones transgénicos, el número de las inclusiones nucleares en las células de Purkinje (Xia et al., 2004). Mientras estos resultados muestran que este tipo de terapia de ARNi en los ensayos preclínicos mejora el comportamiento anómalo característico, su aplicación en pacientes para aliviar o incluso revertir los fenotipos de la enfermedad se retardará hasta que sean evaluadas las pruebas de toxicidad apropiadas.
Apuntando a una diana diferente, las chaperonas moleculares proporcionan una primera línea de defensa contra la mal-plegación y agregación de proteínas. Muchos estudios han analizado los efectos que la sobrexpresión de chaperonas tienen en la formación de cuerpos de inclusión y toxicidad patogénica de fragmentos polyQ en células en cultivo, y está claro que la sobrexpresión molecular de chaperonas podría mostrar ser beneficiosa para el tratamiento de enfermedades neurodegenerativas (Muchowski y Wacker, 2005). Ellas previenen las interacciones impropias dentro de, y entre, polipéptidos no-nativos, reforzando la eficacia de la proteína plegada de novo, y promoviendo el replegado de proteínas que se han mal plegado como resultado de mutaciones y/o del estrés celular (Chan et al., 2000). Las chaperonas químicas y moleculares también podrían prevenir la toxicidad bloqueando las interacciones impropias de la proteína, facilitando la degradación de las proteínas enfermas, o bloqueando eventos de señalización que llevan a la disfunción neuronal y apoptosis. Congo Red, tioflavina S, crisamina G, y Direct Fast, han mostrado eficacia suprimiendo la agregación in vitro y in vivo (Heiser et al., 2000; Sanchez et al., 2003). Algunos compuestos moleculares, como el solvente orgánico dimetilsulfóxido (DMSO) y el celular osmolito glicerol, el trimetilamina N-óxido y trehalosa, aumentan la estabilidad de las proteínas en su estructura nativa; pues son conocidos como chaperonas químicas en base a su influencia en el plegado de la proteína. Son eficaces previniendo la muerte celular activada por la ataxina 3 mutada (Yoshida et al., 2002). La trehalosa fue identificada en una monitorización in vitro como inhibidor de la agregación polyQ, y su administración reduce la atrofia del cerebro, mejora la disfunción motora y extiende la vida de los ratones enfermos con la enfermedad de Huntington (Tanaka et al., 2004). Experimentos in vitro sugieren que los efectos beneficiosos de la trehalosa resultan de su habilidad ligando y estabilizando proteínas que contienen poliglutamina. Más recientemente, se ha identificado en ensayos usando células-libres, una nueva generación de pequeños compuestos químicos dirigidos directamente a la agregación polyQ sin citotoxicidad significativa (Heiser et al., 2002; Zhang et al., 2005). Estos compuestos inhiben la agregación polyQ en células en cultivo y neuronas intactas y podrían eximir, in vivo, de la neurodegeneración causada por polyQ.
Por un mecanismo diferente, una pequeña molécula que actúa como un co-inductor de la respuesta de choque de temperatura prolongando la actividad de factor de transcripción de HSF1, arimoclomol, mejora significativamente los fenotipos de comportamiento, previene la pérdida neuronal, prolonga la supervivencia y ralentiza la progresión de la enfermedad, en un modelo del ratón con neurodegeneración (Kieran et al., 2004). Similarmente, la activación de la respuesta de choque de temperatura con geldanamicina inhibe la agregación y previene la muerte celular (Rimoldi et al., 2001). Esto sugiere que la activación farmacológica de la respuesta celular de choque de temperatura puede, por consiguiente ser un acercamiento terapéutico eficaz para tratar las enfermedades neurodegenerativas.
No obstante, la sobreregulación excesivas de chaperonas puede conducir a unos efectos secundarios indeseables, como alteraciones en la regulación del ciclo celular y cáncer (Mosser y Morimoto, 2004). Por consiguiente, probablemente se requiera un delicado equilibrio en la regulación de chaperonas para obtener un efecto neuroprotector beneficioso. Por ejemplo, las chaperonas químicas o moleculares, usadas en combinación con un agente farmacológico que sobreregule la síntesis de chaperonas moleculares, podría ser un acercamiento terapéutico válido para tratar las ataxias espinocerebelosas causadas por expansiones de poliglutamina. La formación de agregados también ha sido objetivo eficaz con inhibidores de transglutaminasa, como la cistamina que reduce la apoptótica muerte celular y alivia los síntomas de la enfermedad causados por poliglutamina expandida (Dedeoglu et al., 2002; Karpuj et al., 2002).
Los compuestos dirigidos a modular la función mitocondrial como la coenzima Q10 (Shults, 2003), creatina (Ryu et al., 2005) y ácido taurousodeoxicólico (TUDCA) (Keene et al., 2002), o a la autofagia, como el inhibidor mTor rapamicina y su varios análogos (Ravikumar et al., 2004), han mostrado eficacia en la reducción de la toxicidad celular en modelos animales, y actualmente está probándose en ensayos clínicos. La activación de caspasa, la cual normalmente precede a la muerte neuronal y celular puede ser objetivo con inhibidores de caspasa, como zVAD-fmk, CrmA, cistamina, FADD DN, y minociclina. Éstos se han mostrado eficaces para prevenir la progresión de la enfermedad, retrasar el inicio de los síntomas, y extender la supervivencia en varios modelos de ratón neurodegenerativo (Ona et al., 1999; Chen et al., 2000; Lesort et al., 2003). Otros agentes promoviendo la eliminación de proteínas mutadas en el Sistema Nervioso Central con bloqueadores de señalización de Ca2+, como los inhibidores específicos subunidad-NR2B de los receptores N-metil-D-aspartato de glutamato, y bloqueadores del receptor metabotrópico glutamato mGluR5, y del inositol 1,4,5-trisfosfato InsP3R1, pueden ser beneficiosos para el tratamiento de algunos subtipos de ataxia espinocerebelosa. También podrían ensayarse acercamientos terapéuticos por la vía ubiquitina-proteasoma, retardando la agregación de proteínas y reforzando los efectos deletéreos asociados con la degradación de proteínas tóxicas.
El papel que algunas ataxinas juegan en la transcripción y, más importante, la supresión de los efectos citotóxicos mediada por algunos de sus reguladores co-transcripcionales podría usarse para modular los efectos patológicos de las ataxinas mutadas, abriendo camino para nuevas estrategias terapéuticas en el tratamiento de algunas de las SCA's. El reciente progreso investigador en HDAC ha hecho posible el desarrollo de inhibidores específicos de la familia de proteínas HDAC, y estos compuestos podrían ser candidatos eficaces para el tratamiento de la ataxia espinocerebelosa (Dokmanovic y Marks, 2005). Las estrategias neuroprotectoras dirigidas a los defectos específicos bioenergéticos podrían significar una promesa particular en el tratamiento de las condiciones espinocerebelosas. La neuroproteción por el factor de crecimiento insulínico IGF-1 regulando la vía de señalización PI3K/Akt levanta su posible potencial para detener la neurodegeneración cerebelosa (Leinninger y Feldman, 2005).
Las aproximaciones de terapia génica y celular son consideradas para el tratamiento de neurodegeneraciones espinocerebelosas. La diseminación de proteínas o compuestos mediante vectores virales representa una aproximación terapéutica válida. Las terapias celulares de reemplazo neuronal están basadas en la idea de que la función neurológica perdida durante la neurodegeneración podría mejorarse introduciendo nuevas células que puedan formar las conexiones apropiadas y puedan reemplazar la función de las neuronas perdidas. Esta estrategia ha mostrado su eficacia tratando la neurodegeneración en la enfermedad de Parkinson y en la esclerosis lateral amiotrófica (Svendsen y Langston, 2004). En ambos, ratón y humano, las células troncales embrionarias, cuando son cultivadas in vitro para producir cuerpos embrionarios, se diferencian hacia un linaje de la neurona motora (Wichterle et al., 2002). Notablemente, cuando las neuronas, creadas in vitro, son introducidas dentro de la zona dañada del cordón espinal, alguna célula troncal derivada de la neurona motora extiende axones e inervan las correctas dianas. Puesto que la neurogénesis tiene lugar en el sistema nervioso adulto, otro acercamiento podría ser estimular a las células troncales endógenas en el cerebro/cerebelo o en el cordón espinal a generar nuevas neuronas. Los estudios para entender los determinantes moleculares y sugestiones para estimular a las células troncales endógenas están en curso (Gage, 2002). Aunque prometedor, sólo se están empezando a entender los potenciales y desafíos de las células troncales, sobre todo para el uso en las enfermedades neurodegenerativas.
Comentarios de conclusión:
En esta revisión, hemos repasado temas corrientes que tienen lugar en las condiciones neurodegenerativas espinocerebelosas. La neurodegeneración progresiva en las ataxia espinocerebelosas es mediada por proteínas mutadas capaces de inducir el deterioro neuronal interfiriendo con varias rutas moleculares, incluida la agregación y eliminación de proteínas, la regulación transcripcional, el sistema ubiquitina-proteasoma, alteraciones de homeostasis del calcio, y activación de rutas pro-apoptóticas, entre otras. Estas vías podrían actuar independientemente o, más probablemente, interactúar unas con otras, activando la acumulación del deterioro celular que en el futuro lleva a la disfunción y, finalmente, a la muerte de las neuronas a través de una serie de eventos múltiples. Esto sugiere que tratar simultáneamente dianas en varias rutas moleculares pudiera ser terapéuticamente necesario para prevenir la neurodegeneración y conservar la función neuronal. Entender cómo la desregulación de estas vías media en la progresión de la enfermedad está llevando al establecimiento de estrategias terapéuticas eficaces in vivo, las cuales pudieran mostrarse beneficiosas en el tratamiento de las ataxias espinocerebelosas.
Reconocimientos:
Estamos agradecidos con Nick Wood (Institute of Neurology, Queen Square, Londres, Reino Unido) por su apoyo, comentarios útiles y sugerencias. Este trabajo fue apoyado por el Child Health Research Appeal Trust (CHRAT) y el VI Programa Marco (FP6) de la Comisión Europea (EUROSCA Integrated Project LHSM-CT-2004-503304). La investigación en el Institute of Child Health (ICH) se beneficia en Investigación y Desarrollo subvencionado desde el National Health System Executive, del Reino Unido.
Referencias:
Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S. PKC gamma mutant mice exhibit mild deficits in spatial and contextual learning. Cell 1993; 75: 1263-71.
Albrecht M, Golatta M, Wullner U, Lengauer T. Structural and functional analysis of ataxin-2 and ataxin-3. Eur J Biochem 2004; 271: 3155-70.
Bonini NM. Chaperoning brain degeneration. Proc Natl Acad Sci USA 2002; 99: 16407-11.
Buhmann C, Bussopulos A, Oechsner M. Dopaminergic response in parkinsonian phenotype of Machado-Joseph disease. Mov Disord 2003; 18: 219-21.
Cagnoli C, Mariotti C, Taroni F, Seri M, Brussino A, Michielotto C, et al. SCA28, a novel form of autosomal dominant cerebellar ataxia on chromosome 18p11.22-q11.2. Brain 2006; 129: 235-42.
Chai Y, Berke SS, Cohen RE, Paulson HL. Poly-ubiquitin binding by the polyglutamine disease protein ataxin-3 links its normal function to protein surveillance pathways. J Biol Chem 2004; 279: 3605-11.
Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet 2000; 9: 2811-20.
Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 2000; 6: 797-801.
Chen DH, Brkanac Z, Verlinde CL, Tan XJ, Bylenok L, Nochlin D, et al. Missense mutations in the regulatory domain of PKCgamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet 2003a; 72: 839-49.
Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 2003b; 113: 457-68.
Chou AH, Yeh TH, Kuo YL, Kao YC, Jou MJ, Hsu CY, et al. Polyglutamine-expanded ataxin-3 activates mitochondrial apoptotic pathway by upregulating Bax and downregulating Bcl-x(L). Neurobiol Dis 2006; 21: 333-45.
Chow MK, Ellisdon AM, Cabrita LD, Bottomley SP. Polyglutamine expansion in ataxin-3 does not affect protein stability: implications for misfolding and disease. J Biol Chem 2004; 279: 47643-51.
Chung MY, Lu YC, Cheng NC, Soong BW. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain 2003; 126: 1293-9.
Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci 1997; 17: 7385-95.
Coates JC. Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol 2003; 13: 463-71.
Corral J, Genis D, Banchs I, San Nicolas H, Armstrong J, Volpini V. Giant SCA8 alleles in nine children whose mother has two moderately large ones. Ann Neurol 2005; 57: 549-53.
Craig K, Keers SM, Archibald K, Curtis A, Chinnery PF. Molecular epidemiology of spinocerebellar ataxia type 6. Ann Neurol 2004; 55: 752-5.
Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 1998; 19: 148-54.
Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, et al. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 1999; 24: 879-92.
Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, et al. Overexpression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet 2001; 10: 1511-8.
David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 1997; 17: 65-70.
Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, et al. Therapeutic effects of cystamine in a murine model of Huntington's disease. J Neurosci 2002; 22: 8942-50.
Devos D, Schraen-Maschke S, Vuillaume I, Dujardin K, Naze P, Willoteaux C, et al. Clinical features and genetic analysis of a new form of spinocerebellar ataxia. Neurol 2001; 56: 234-8.
Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem 2005; 96: 293-304.
Flanigan K, Gardner K, Alderson K, Galster B, Otterud B, Leppert MF, et al. Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy (SCA4): clinical description and genetic localization to chromosome 16q22.1. Am J Hum Genet 1996; 59: 392-9.
Gage FH. Neurogenesis in the adult brain. J Neurosci 2002; 22: 612-3.
Gardner RJ, Knight MA, Hara K, Tsuji S, Forrest SM, Storey E. Spinocerebellar ataxia type 15. Cerebellum 2005; 4: 47-50.
Gazulla J, Errea JM, Benavente I, Tordesillas CJ. Treatment of ataxia in cortical cerebellar atrophy with the GABAergic drug gabapentin. A preliminary study. Eur Neurol 2004; 52: 7-11.
Gilman S, Sima AA, Junck L, Kluin KJ, Koeppe RA, Lohman ME, et al. Spinocerebellar ataxia type 1 with multiple system degeneration and glial cytoplasmic inclusions. Ann Neurol 1996; 39: 241-55.
Giunti P, Stevanin G, Worth PF, David G, Brice A, Wood NW. Molecular and clinical study of 18 families with ADCA type II: evidence for genetic heterogeneity and de novo mutation. Am J Hum Genet 1999; 64: 1594-603.
Green H. Human genetic diseases due to codon reiteration: relationship to an evolutionary mechanism. Cell 1993; 74: 955-6.
Gunther P, Storch A, Schwarz J, Sabri O, Steinbach P, Wagner A, et al. Basal ganglia involvement of a patient with SCA 17-a new form of autosomal dominant spinocerebellar ataxia. J Neurol 2004; 251: 896-7.
Gwinn-Hardy K, Singleton A, O'Suilleabhain P, Boss M, Nicholl D, Adam A, et al. Spinocerebellar ataxia type 3 phenotypically resembling Parkinson disease in a black family. Arch Neurol 2001; 58: 296-9.
Harding AE. Clinical features and classification of inherited ataxias. In: Harding AE, Deufel T, editors. Inherited ataxias. Vol 61. New York: Raven; 1993. p. 1-14.
He W, Parker R. Functions of Lsm proteins in mRNA degradation and splicing. Curr Opin Cell Biol 2000; 12: 346-50. Molecular pathogenesis of spinocerebellar ataxias Brain (2006) Page 11 of 14.
Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, et al. Inhibition of Huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington's disease therapy. Proc Natl Acad Sci USA 2000; 97: 6739-44.
Heiser V, Engemann S, Brocker W, Dunkel I, Boeddrich A, Waelter S, et al. Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington's disease by using an automated filter retardation assay. Proc Natl Acad Sci USA 2002; 99: 16400-6.
Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet 2004; 13: 1257-65.
Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA 2003; 100: 2041-6.
Hoffner G, Djian P. Transglutaminase and diseases of the central nervous system. Front Biosci 2005; 10: 3078-92.
Holmes SE, O'Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, et al. Expansion of a novel CAG trinucleotide repeat in the 50 region of PPP2R2B is associated with SCA12. Nat Genet 1999; 23: 391-2.
Ichise T, Kano M, Hashimoto K, Yanagihara D, Nakao K, Shigemoto R, et al. mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science 2000; 288: 1832-5.
Igarashi S, Koide R, Shimohata T, Yamada M, Hayashi Y, Takano H, et al. Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA protein with an expanded polyglutamine stretch. Nat Genet 1998; 18: 111-7.
Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet 2006; 38: 184-90.
Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 1996; 14: 285-91.
Infante J, Berciano J, Volpini V, Corral J, Polo JM, Pascual J, et al. Spinocerebellar ataxia type 2 with Levodopa-responsive parkinsonism culminating in motor neuron disease. Mov Disord 2004; 19: 848-52.
Ishikawa K, Toru S, Tsunemi T, Li M, Kobayashi K, Yokota T, et al. An autosomal dominant cerebellar ataxia linked to chromosome 16q22.1 is associated with a single-nucleotide substitution in the 50 untranslated region of the gene encoding a protein with spectrin repeat and Rho guanine-nucleotide exchange-factor domains. Am J Hum Genet 2005; 77: 280-96.
Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, et al. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci USA 2005; 102: 13135-40.
Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, et al. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J 2003; 22: 905-12.
Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, et al. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med 2002; 8: 143-9.
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 1994; 8: 221-7.
Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc Natl Acad Sci USA 2002; 99: 10671-6.
Khan NL, Giunti P, Sweeney MG, Scherfler C, Brien MO, Piccini P, et al. Parkinsonism and nigrostriatal dysfunction are associated with spinocerebellar ataxia type 6 (SCA6). Mov Disord 2005; 20: 1115-9.
Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 2004; 10: 402-5.
Knight MA, Gardner RJ, Bahlo M, Matsuura T, Dixon JA, Forrest SM, et al. Dominantly inherited ataxia and dysphonia with dentate calcification. spinocerebellar ataxia type 20. Brain 2004; 127: 1172-81.
Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet 1994; 6: 9-13.
Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 1999; 21: 379-84.
Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J 2000; 19: 1176-9.
La Spada AR, Fu Y, Sopher BL, Libby RT, Wang X, Li LY, et al. Polyglutamine-expanded ataxin-7 antagonizes crx function and induces cone-rod dystrophy in a mouse model of sca7. Neuron 2001; 31: 913-27.
Leinninger GM, Feldman EL. Insulin-like growth factors in the treatment of neurological disease. Endocr Dev 2005; 9: 135-59. Lesort M, Lee M, Tucholski J, Johnson GV. Cystamine inhibits caspase activity. Implications for the treatment of polyglutamine disorders. J Biol Chem 2003; 278: 3825-30.
Li F, Macfarlan T, Pittman RN, Chakravarti D. Ataxin-3 is a histone-binding protein with two independent transcripcional corepressor activities. J Biol Chem 2002; 277: 45004-12.
Lim J, Lu KP. Pinning down phosphorylated tau and tauopathies. Biochem Biophys Acta 2005; 1739: 311-22.
Lin X, Ashizawa T. Recent progress in spinocerebellar ataxia type-10 (SCA10). Cerebellum 2005; 4: 37-42.
Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci 2000; 3: 157-63.
Lipinski MM, Yuan J. Mechanisms of cell death in polyglutamine expansion diseases. Curr Opin Pharmacol 2004; 4: 85-90.
Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001; 293: 864-7.
Mantuano E, Veneziano L, Jodice C, Frontali M. Spinocerebellar ataxia type 6 and episodic ataxia type 2: differences and similarities between two allelic disorders. Cytogenet Genome Res 2003; 100: 147-53.
Matilla A, Radrizzani M. The Anp32 family of proteins containing leucine-rich repeats. Cerebellum 2005; 4: 7-18.
Matilla A, Koshy BT, Cummings CJ, Isobe T, Orr HT, Zoghbi HY. The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature 1997; 389: 974-8.
Matilla A, Gorbea C, Einum DD, Townsend JJ, Michalik A, van Broeckhaven C, et al. Association of ataxin-7 with the proteasome subunit S4 of the 19S regulatory complex. Hum Mol Genet 2001; 10: 2821-31.
Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 2000; 26: 191-4.
Matsuyama Z, Yanagisawa NK, Aoki Y, Black JL III, Lennon VA, Mori Y, et al. Polyglutamine repeats of spinocerebellar ataxia 6 impair the cell-deathpreventing effect of CaV2.1 Ca2+ channel-loss-of-function cellular model of SCA6. Neurobiol Dis 2004; 17: 198-204.
McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, et al. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 2000; 9: 2197-202.
McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci USA 2001; 98: 15179-84.
Miyoshi Y, Yamada T, Tanimura M, Taniwaki T, Arakawa K, Ohyagi Y, et al. A novel autosomal dominant spinocerebellar ataxia (SCA16) linked to chromosome 8q22.1-24.1. Neurol 2001; 57: 96-100.
Mizutani A, Wang L, Rajan H, Vig PJ, Alaynick WA, Thaler JP, et al. Boat, an AXH domain protein, suppresses the cytotoxicity of mutant ataxin-1. EMBO J 2005; 24: 3339-51.
Mosser DD, Morimoto RI. Molecular chaperones and the stress of oncogenesis. Oncogene 2004; 23: 2907-18.
Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 2005; 6: 11-22.
Mutsuddi M, Marshall CM, Benzow KA, Koob MD, Rebay I. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr Biol 2004; 14: 302-8.
Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, Bundo M, et al. Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat Genet 1994; 6: 14-18.
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 2001; 10: 1441-8.
Nandagopal R, Moorthy SG. Dramatic levodopa responsiveness of dystonia in a sporadic case of spinocerebellar ataxia type 3. Postgrad Med J 2004; 80: 363-5.
Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 2001; 101: 2353-64.
Nicastro G, Menon RP, Masino L, Knowles PP, McDonald NQ, Pastore A. The solution structure of the Josephin domain of ataxin-3: structural determinants for molecular recognition. Proc Natl Acad Sci USA 2005; 102: 10493-8.
Okazawa H, Rich T, Chang A, Lin X, Waragai M, Kajikawa M, et al. Interaction between mutant ataxin-1 and PQBP-1 affects transcripcion and cell death. Neuron 2002; 34: 701-13.
Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 1999; 399: 263-7.
Orr H, Chung M-Y, Banfi S, Kwiatkowski Jr TJ, Servadio A, Beaudet al, et al. Expansion of an unstable trinucleotide (CAG) repeat in spinocerebellar ataxia type 1. Nat Genet 1993; 4: 221-6.
Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 2003; 4: 552-65.
Park Y, Hong S, Kim SJ, Kang S. Proteasome function is inhibited by polyglutamine-expanded ataxin-1, the SCA1 gene product. Mol Cells 2005; 19: 23-30.
Petersen OH. Calcium signal compartmentalization. Biol Res 2002; 35: 177-82.
Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998; 280: 737-41.
Pietrobon D. Calcium channels and channelopathies of the central nervous system. Mol Neurobiol 2002; 25: 31-50.
Pirker W, Back C, Gerschlager W, Laccone F, Alesch F. Chronic thalamic stimulation in a patient with spinocerebellar ataxia type 2. Mov Disord 2003; 18: 222-5.
Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996; 14: 269-76.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585-95.
Rimoldi M, Servadio A, Zimarino V. Analysis of heat shock transcripcion factor for suppression of polyglutamine toxicity. Brain Res Bull 2001; 56: 353-62.
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med 2004; 10: S10-7.
Ryu H, Rosas HD, Hersch SM, Ferrante RJ. The therapeutic role of creatine in Huntington's disease. Pharmacol Ther 2005; 108: 193-207.
Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc Natl Acad Sci USA 2002; 99: 16412-8.
Sanchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003; 421: 373-9.
Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 1996; 14: 277-84.
Schmidt T, Lindenberg KS, Krebs A, Schols L, Laccone F, Herms J, et al. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann Neurol 2002; 51: 302-10.
Schols L, Peters S, Szymanski S, Kruger R, Lange S, Hardt C, et al. Extrapyramidal motor signs in degenerative ataxias. Arch Neurol 2000; 57: 1495-500.
Schrenk K, Kapfhammer JP, Metzger F. Altered dendritic development of cerebellar Purkinje cells in slice cultures from protein kinase Cgammadeficient mice. Neuroscience 2002; 110: 675-89.
Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet 2004; 13: 2535-43.
Shibata H, Huynh DP, Pulst SM. A novel protein with RNA-binding motifs interacts with ataxin-2. Hum Mol Genet 2000; 9: 1303-13.
Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, et al. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcripcion. Nat Genet 2000; 26: 29-36.
Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306: 990-5.
Shults CW. Coenzyme Q10 in neurodegenerative diseases. Curr Med Chem 2003; 10: 1917-21.
Simon-Sanchez J, Hanson M, Singleton A, Hernandez D, McInerney A, Nussbaum R, et al. Analysis of SCA-2 and SCA-3 repeats in parkinsonism: evidence of SCA-2 expansion in a family with autosomal dominant Parkinson's disease. Neurosci Lett 2005; 382: 191-4.
Sontag E. Protein phosphatase 2A: the Trojan Horse of cellular signaling. Cell Signal 2001; 13: 7-16.
Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001; 413: 739-43.
Stevanin G, Broussolle E, Streichenberger N, Kopp N, Brice A, Durr A. Spinocerebellar ataxia with sensory neuropathy (SCA25). Cerebellum 2005; 4: 58-61.
Sun XM, Butterworth M, MacFarlane M, Dubiel W, Ciechanover A, Cohen GM. Caspase activation inhibits proteasome function during apoptosis. Mol Cell 2004; 14: 81-93.
Svendsen CN, Langston JW. Stem cells for Parkinson disease and ALS: replacement or protection? Nat Med 2004; 10: 224-5.
Swartz BE, Burmeister M, Somers JT, Rottach KG, Bespalova IN, Leigh RJ. A form of inherited cerebellar ataxia with saccadic intrusions, increased saccadic speed, sensory neuropathy, and myoclonus. Ann NY Acad Sci 2002; 956: 441-4.
Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 2004; 10: 148-54.
Tsai CC, Kao HY, Mitzutani A, Banayo E, Rajan H, McKeown M, et al. Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci USA 2004; 101: 4047-52.
Tsuda H, Jafar-Nejad H, Patel AJ, Sun Y, Chen HK, Rose MF, et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell 2005; 122: 633-44.
van de Warrenburg B, Sinke R, Verschuuren-Bemelmans C, Scheffer H, Brunt E, Ippel P, et al. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis. Neurol 2002; 58: 702-8.
van de Warrenburg BP, Hendriks H, Durr A, van Zuijlen MC, Stevanin G, Camuzat A, et al. Age at onset variance analysis in spinocerebellar ataxias: a study in a Dutch-French cohort. Ann Neurol 2005; 57: 505-12.
van Swieten JC, Brusse E, de Graaf BM, Krieger E, van de Graaf R, de Koning I, et al. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebral ataxia. Am J Hum Genet 2003; 72: 191-9.